作者:中华医学会 中华医学会杂志社 中华医学会全科医学分会 中华医学会呼吸病学分会慢阻肺学组 中华医学会《中华全科医师杂志》编辑委员会 呼吸系统疾病基层诊疗指南编写专家组
本文刊于:中华全科医师杂志, 2018,17(11) : 856-870
作者:中华医学会 中华医学会杂志社 中华医学会全科医学分会 中华医学会呼吸病学分会慢阻肺学组 中华医学会《中华全科医师杂志》编辑委员会 呼吸系统疾病基层诊疗指南编写专家组
本文刊于:中华全科医师杂志, 2018,17(11) : 856-870
一. 概述
(一)定义
慢性阻塞性肺疾病(chronic obstructive pulmonary disease,COPD)简称慢阻肺,是一种以持续气流受限为特征的可以预防和治疗的常见疾病,气流受限多呈进行性发展,与气道和肺对有毒颗粒或气体的慢性炎症反应增强有关。急性加重和合并症对个体患者整体疾病的严重程度产生影响。慢性气流受限由小气道疾病(阻塞性支气管炎)和肺实质破坏(肺气肿)共同引起,两者在不同患者所占比重不同[1]。
通常,慢性支气管炎是指在除外慢性咳嗽的其他已知病因后,患者每年咳嗽、咳痰3个月以上,并连续2年以上者。肺气肿则是指肺部终末细支气管远端气腔出现异常持久的扩张,并伴有肺泡壁和细支气管破坏而无明显的肺纤维化。当慢性支气管炎和肺气肿患者的肺功能检査出现持续气流受限时,则可诊断为慢阻肺;如患者仅有"慢性支气管炎"和/或"肺气肿",而无持续气流受限,则不能诊断为慢阻肺[1]。
虽然支气管哮喘(哮喘)与慢阻肺都是慢性气道炎症性疾病,但二者的发病机制不同,临床表现及对治疗的反应性也有明显差别。大多数哮喘患者的气流受限具有显著的可逆性,这是其不同于慢阻肺的一个关键特征。但是,部分哮喘患者随着病程延长,可出现较明显的气道重塑,导致气流受限的可逆性明显减小,临床很难与慢阻肺相鉴别。慢阻肺和哮喘可以发生于同一患者,且由于两者都是常见病、多发病,这种概率并不低[1]。
一些已知病因或具有特征性病理表现的气流受限疾病,如支气管扩张症、肺结核、弥漫性泛细支气管炎和闭塞性细支气管炎等均不属于慢阻肺。
(二)流行病学
慢阻肺是一种严重危害人类健康的常见病和多发病,严重影响患者的生命质量,病死率较高,并给患者及其家庭以及社会带来沉重的经济负担。2007年对我国7个地区20 245名成年人的调査结果显示,40岁以上人群中慢阻肺的患病率高达8.2%[2]。2018年中国成人肺部健康研究(CPHS)对10个省市50 991名人群调查显示20岁及以上成人的慢阻肺患病率为8.6%,40岁以上则高达13.7%,首次明确我国慢阻肺患者人数近1亿,慢阻肺已经成为与高血压、糖尿病"等量齐观"的慢性疾病,构成重大疾病负担[3]。据统计2013年中国慢阻肺死亡人数约91.1万人,占全世界慢阻肺死亡人数的1/3[4],远高于中国肺癌年死亡人数。据全球疾病负担研究项目估计,2020年慢阻肺将位居全球死亡原因的第3位[5,6]。世界银行/WHO的资料表明,至2020年慢阻肺将位居世界疾病经济负担的第5位[7]。
(三)分期[1]
1.急性加重期:
患者呼吸道症状加重,超过日常变异水平,需要改变治疗方案。表现为咳嗽、咳痰、气短和/或喘息加重,痰量增多,脓性或黏液脓性痰,可伴有发热等。
2.稳定期:
咳嗽、咳痰和气短等症状稳定或症状轻微,病情基本恢复到急性加重前的状态。
(一)定义
慢性阻塞性肺疾病(chronic obstructive pulmonary disease,COPD)简称慢阻肺,是一种以持续气流受限为特征的可以预防和治疗的常见疾病,气流受限多呈进行性发展,与气道和肺对有毒颗粒或气体的慢性炎症反应增强有关。急性加重和合并症对个体患者整体疾病的严重程度产生影响。慢性气流受限由小气道疾病(阻塞性支气管炎)和肺实质破坏(肺气肿)共同引起,两者在不同患者所占比重不同[1]。
通常,慢性支气管炎是指在除外慢性咳嗽的其他已知病因后,患者每年咳嗽、咳痰3个月以上,并连续2年以上者。肺气肿则是指肺部终末细支气管远端气腔出现异常持久的扩张,并伴有肺泡壁和细支气管破坏而无明显的肺纤维化。当慢性支气管炎和肺气肿患者的肺功能检査出现持续气流受限时,则可诊断为慢阻肺;如患者仅有"慢性支气管炎"和/或"肺气肿",而无持续气流受限,则不能诊断为慢阻肺[1]。
虽然支气管哮喘(哮喘)与慢阻肺都是慢性气道炎症性疾病,但二者的发病机制不同,临床表现及对治疗的反应性也有明显差别。大多数哮喘患者的气流受限具有显著的可逆性,这是其不同于慢阻肺的一个关键特征。但是,部分哮喘患者随着病程延长,可出现较明显的气道重塑,导致气流受限的可逆性明显减小,临床很难与慢阻肺相鉴别。慢阻肺和哮喘可以发生于同一患者,且由于两者都是常见病、多发病,这种概率并不低[1]。
一些已知病因或具有特征性病理表现的气流受限疾病,如支气管扩张症、肺结核、弥漫性泛细支气管炎和闭塞性细支气管炎等均不属于慢阻肺。
(二)流行病学
慢阻肺是一种严重危害人类健康的常见病和多发病,严重影响患者的生命质量,病死率较高,并给患者及其家庭以及社会带来沉重的经济负担。2007年对我国7个地区20 245名成年人的调査结果显示,40岁以上人群中慢阻肺的患病率高达8.2%[2]。2018年中国成人肺部健康研究(CPHS)对10个省市50 991名人群调查显示20岁及以上成人的慢阻肺患病率为8.6%,40岁以上则高达13.7%,首次明确我国慢阻肺患者人数近1亿,慢阻肺已经成为与高血压、糖尿病"等量齐观"的慢性疾病,构成重大疾病负担[3]。据统计2013年中国慢阻肺死亡人数约91.1万人,占全世界慢阻肺死亡人数的1/3[4],远高于中国肺癌年死亡人数。据全球疾病负担研究项目估计,2020年慢阻肺将位居全球死亡原因的第3位[5,6]。世界银行/WHO的资料表明,至2020年慢阻肺将位居世界疾病经济负担的第5位[7]。
(三)分期[1]
1.急性加重期:
患者呼吸道症状加重,超过日常变异水平,需要改变治疗方案。表现为咳嗽、咳痰、气短和/或喘息加重,痰量增多,脓性或黏液脓性痰,可伴有发热等。
2.稳定期:
咳嗽、咳痰和气短等症状稳定或症状轻微,病情基本恢复到急性加重前的状态。
二. 病因与发病机制
(一)危险因素
慢阻肺的发病是遗传与环境因素共同作用的结果。
1.遗传因素:
某些遗传因素可增加慢阻肺发病的危险,即慢阻肺有遗传易感性。已知的遗传因素为α1-抗胰蛋白酶缺乏[8]。α1-抗胰蛋白酶是一种蛋白酶抑制剂,重度α1-抗胰蛋白酶缺乏与非吸烟者的肺气肿形成有关。在我国,α1-抗胰蛋白酶缺乏引起的肺气肿迄今尚未见正式报道。
2.吸烟:
吸烟是慢阻肺最重要的环境发病因素。吸烟者的肺功能异常率较高,第1秒用力呼气容积(forced expiratory volume in one second,FEV1)年下降率更快,吸烟者死于慢阻肺的人数多于非吸烟者[9]。但并非所有的吸烟者均发展成具有显著临床症状的慢阻肺。被动吸烟也可能导致呼吸道症状及慢阻肺的发生[10]。孕妇吸烟可能会影响胎儿肺脏的生长及其在子宫内的发育,并对胎儿的免疫系统功能产生一定影响[11]。
3.空气污染:
空气中的烟尘或二氧化硫明显增加时,慢阻肺急性加重显著增多。其他粉尘也能刺激支气管黏膜,使气道清除功能遭受损害,为细菌入侵创造条件[1]。研究表明大气中直径2.5~10 μm的颗粒物(particulate matter,PM),即PM 2.5、PM10水平的升高与慢阻肺的发生显著增加相关[12]。木材、动物粪便、农作物残梗、煤炭等,以明火或在通风功能不佳的火炉中燃烧,可导致严重的室内空气污染,是导致慢阻肺的重要危险因素[1]。
4.职业性粉尘和化学物质:
当职业性粉尘及化学物质(烟雾、过敏原、有机与粉尘、工业废气等)的浓度过大或接触时间过久,均可导致慢阻肺的发生。接触某些特殊物质、刺激性物质、有机粉尘及过敏原也可使气道反应性增加[1]。
5.感染:
呼吸道感染是慢阻肺发病和急性加重的另一个重要因素,病毒和/或细菌感染与气道炎症加剧有关[13],是慢阻肺急性加重的常见原因。儿童期有严重的呼吸道感染史与成年时肺功能降低及呼吸系统症状的发生有关[1]。
6.社会经济地位:
慢阻肺的发生风险与患者的社会经济地位呈负相关[14]。这可能与低社会经济状态与室内及室外空气污染暴露、拥挤、营养状态差或其他因素有关[15]。
(二)发病机制
慢阻肺的发病机制尚未完全明确,肺部炎症反应、氧化应激、蛋白酶和抗蛋白酶失衡等在慢阻肺的发病中起重要作用。
1.慢性炎症反应:
慢阻肺主要以外周气道、肺实质和肺血管中增加的巨噬细胞为特征,同时还伴有活化的中性粒细胞和淋巴细胞,后者包括细胞毒性T细胞(Tc)1、辅助性T细胞(Th)1、Th17、固有淋巴细胞(ILC)3。急性加重期较稳定期炎症反应更为明显。一些患者也可能出现嗜酸粒细胞、Th 2或ILC 2细胞增加,尤其是临床上和哮喘有重叠时。所有这些炎症细胞和上皮细胞及其他结构细胞一起释放多种炎症介质[16]。炎症介质水平增高,吸引循环中的炎症细胞,放大炎症过程,诱导结构改变[17]。
2.氧化应激:
氧化应激可能是慢阻肺重要的炎症放大机制[16,18]。氧化应激的生物标志物(如过氧化氢,8-异前列腺素)在慢阻肺患者呼出气冷凝液、痰、体循环中浓度升高。慢阻肺急性加重时,氧化应激进一步加重。氧化剂由香烟及其他吸入颗粒刺激产生,并通过巨噬细胞和中性粒细胞等活化的炎症细胞释放出来。慢阻肺患者也可能会存在内源性抗氧化剂的减少,这源于核因子2(Nrf 2)转录因子的减少,它参与调节多种抗氧化基因[19,20]。
3.蛋白酶-抗蛋白酶失衡:
已有证据表明慢阻肺患者肺组织中蛋白酶与抗蛋白酶表达失衡,前者可降解结缔组织,后者与之相反[21]。蛋白酶介导弹性蛋白的破坏,后者是肺实质中重要的结缔组织成分,这种破坏是肺气肿的重要特征[22]。
4.细支气管周围和间质纤维化:
慢阻肺患者或无症状吸烟者中存在细支气管周围纤维化和间质改变[23,24,25,26]。吸烟者或有气道炎症的慢阻肺患者中发现有过量的生长因子产生[27],炎症可先于纤维化发生,或气道壁反复损伤本身导致肌纤维组织过度产生[28],从而促进小气道气流受限的发生,最终导致气道闭塞,继发肺气肿[29]。
(三)病理生理[1,15]
慢阻肺的特征性病理改变表现在气道、肺实质及肺血管[30],肺组织不同部位出现以特异性炎症细胞增多为特征的慢性炎症,以及反复损伤与修复后出现的结构改变。修复过程导致气道壁结构重构,胶原含量增加及瘢痕组织形成,这些病理改变造成气道狭窄,引起固定性气道阻塞。
在慢阻肺的肺部病理学改变基础上,出现相应的慢阻肺特征性病理生理学改变,包括黏液高分泌、纤毛功能失调、小气道炎症、纤维化及管腔内渗出、气流受限、肺过度充气、气体交换异常、肺动脉高压和肺源性心脏病以及全身的不良效应。黏液高分泌和纤毛功能失调导致慢性咳嗽及多痰,这些症状可出现在其他症状和病理生理异常发生之前。随着慢阻肺的进展,外周气道阻塞、肺实质破坏及肺血管异常等降低了肺气体交换能力,产生低氧血症,并可出现高碳酸血症。长期慢性缺氧可导致肺血管广泛收缩和肺动脉高压,常伴有血管内膜增生,某些血管发生纤维化和闭塞,导致肺循环的结构重构。慢阻肺晚期出现肺动脉高压是其重要的心血管并发症,进而导致慢性肺源性心脏病及右心衰竭,提示预后不良。
慢阻肺的炎症反应不仅局限于肺部,也可以导致全身不良效应。全身炎症反应表现为全身氧化负荷异常增高、循环血液中促炎细胞因子浓度异常增高及炎性细胞异常活化等,进而导致骨质疏松、抑郁、慢性贫血及心血管疾病风险增加。这些可使患者的活动能力受限加剧,生命质量下降,预后变差,因此具有重要的临床意义。
(一)危险因素
慢阻肺的发病是遗传与环境因素共同作用的结果。
1.遗传因素:
某些遗传因素可增加慢阻肺发病的危险,即慢阻肺有遗传易感性。已知的遗传因素为α1-抗胰蛋白酶缺乏[8]。α1-抗胰蛋白酶是一种蛋白酶抑制剂,重度α1-抗胰蛋白酶缺乏与非吸烟者的肺气肿形成有关。在我国,α1-抗胰蛋白酶缺乏引起的肺气肿迄今尚未见正式报道。
2.吸烟:
吸烟是慢阻肺最重要的环境发病因素。吸烟者的肺功能异常率较高,第1秒用力呼气容积(forced expiratory volume in one second,FEV1)年下降率更快,吸烟者死于慢阻肺的人数多于非吸烟者[9]。但并非所有的吸烟者均发展成具有显著临床症状的慢阻肺。被动吸烟也可能导致呼吸道症状及慢阻肺的发生[10]。孕妇吸烟可能会影响胎儿肺脏的生长及其在子宫内的发育,并对胎儿的免疫系统功能产生一定影响[11]。
3.空气污染:
空气中的烟尘或二氧化硫明显增加时,慢阻肺急性加重显著增多。其他粉尘也能刺激支气管黏膜,使气道清除功能遭受损害,为细菌入侵创造条件[1]。研究表明大气中直径2.5~10 μm的颗粒物(particulate matter,PM),即PM 2.5、PM10水平的升高与慢阻肺的发生显著增加相关[12]。木材、动物粪便、农作物残梗、煤炭等,以明火或在通风功能不佳的火炉中燃烧,可导致严重的室内空气污染,是导致慢阻肺的重要危险因素[1]。
4.职业性粉尘和化学物质:
当职业性粉尘及化学物质(烟雾、过敏原、有机与粉尘、工业废气等)的浓度过大或接触时间过久,均可导致慢阻肺的发生。接触某些特殊物质、刺激性物质、有机粉尘及过敏原也可使气道反应性增加[1]。
5.感染:
呼吸道感染是慢阻肺发病和急性加重的另一个重要因素,病毒和/或细菌感染与气道炎症加剧有关[13],是慢阻肺急性加重的常见原因。儿童期有严重的呼吸道感染史与成年时肺功能降低及呼吸系统症状的发生有关[1]。
6.社会经济地位:
慢阻肺的发生风险与患者的社会经济地位呈负相关[14]。这可能与低社会经济状态与室内及室外空气污染暴露、拥挤、营养状态差或其他因素有关[15]。
(二)发病机制
慢阻肺的发病机制尚未完全明确,肺部炎症反应、氧化应激、蛋白酶和抗蛋白酶失衡等在慢阻肺的发病中起重要作用。
1.慢性炎症反应:
慢阻肺主要以外周气道、肺实质和肺血管中增加的巨噬细胞为特征,同时还伴有活化的中性粒细胞和淋巴细胞,后者包括细胞毒性T细胞(Tc)1、辅助性T细胞(Th)1、Th17、固有淋巴细胞(ILC)3。急性加重期较稳定期炎症反应更为明显。一些患者也可能出现嗜酸粒细胞、Th 2或ILC 2细胞增加,尤其是临床上和哮喘有重叠时。所有这些炎症细胞和上皮细胞及其他结构细胞一起释放多种炎症介质[16]。炎症介质水平增高,吸引循环中的炎症细胞,放大炎症过程,诱导结构改变[17]。
2.氧化应激:
氧化应激可能是慢阻肺重要的炎症放大机制[16,18]。氧化应激的生物标志物(如过氧化氢,8-异前列腺素)在慢阻肺患者呼出气冷凝液、痰、体循环中浓度升高。慢阻肺急性加重时,氧化应激进一步加重。氧化剂由香烟及其他吸入颗粒刺激产生,并通过巨噬细胞和中性粒细胞等活化的炎症细胞释放出来。慢阻肺患者也可能会存在内源性抗氧化剂的减少,这源于核因子2(Nrf 2)转录因子的减少,它参与调节多种抗氧化基因[19,20]。
3.蛋白酶-抗蛋白酶失衡:
已有证据表明慢阻肺患者肺组织中蛋白酶与抗蛋白酶表达失衡,前者可降解结缔组织,后者与之相反[21]。蛋白酶介导弹性蛋白的破坏,后者是肺实质中重要的结缔组织成分,这种破坏是肺气肿的重要特征[22]。
4.细支气管周围和间质纤维化:
慢阻肺患者或无症状吸烟者中存在细支气管周围纤维化和间质改变[23,24,25,26]。吸烟者或有气道炎症的慢阻肺患者中发现有过量的生长因子产生[27],炎症可先于纤维化发生,或气道壁反复损伤本身导致肌纤维组织过度产生[28],从而促进小气道气流受限的发生,最终导致气道闭塞,继发肺气肿[29]。
(三)病理生理[1,15]
慢阻肺的特征性病理改变表现在气道、肺实质及肺血管[30],肺组织不同部位出现以特异性炎症细胞增多为特征的慢性炎症,以及反复损伤与修复后出现的结构改变。修复过程导致气道壁结构重构,胶原含量增加及瘢痕组织形成,这些病理改变造成气道狭窄,引起固定性气道阻塞。
在慢阻肺的肺部病理学改变基础上,出现相应的慢阻肺特征性病理生理学改变,包括黏液高分泌、纤毛功能失调、小气道炎症、纤维化及管腔内渗出、气流受限、肺过度充气、气体交换异常、肺动脉高压和肺源性心脏病以及全身的不良效应。黏液高分泌和纤毛功能失调导致慢性咳嗽及多痰,这些症状可出现在其他症状和病理生理异常发生之前。随着慢阻肺的进展,外周气道阻塞、肺实质破坏及肺血管异常等降低了肺气体交换能力,产生低氧血症,并可出现高碳酸血症。长期慢性缺氧可导致肺血管广泛收缩和肺动脉高压,常伴有血管内膜增生,某些血管发生纤维化和闭塞,导致肺循环的结构重构。慢阻肺晚期出现肺动脉高压是其重要的心血管并发症,进而导致慢性肺源性心脏病及右心衰竭,提示预后不良。
慢阻肺的炎症反应不仅局限于肺部,也可以导致全身不良效应。全身炎症反应表现为全身氧化负荷异常增高、循环血液中促炎细胞因子浓度异常增高及炎性细胞异常活化等,进而导致骨质疏松、抑郁、慢性贫血及心血管疾病风险增加。这些可使患者的活动能力受限加剧,生命质量下降,预后变差,因此具有重要的临床意义。
三. 诊断、评估与转诊
(一)慢阻肺高危人群
符合以下1个及以上特征的人群均属于慢阻肺的高危人群:
1.年龄≥35岁。
2.吸烟或长期接触"二手烟"污染。
3.患有某些特定疾病,如支气管哮喘、过敏性鼻炎、慢性支气管炎、肺气肿等。
4.直系亲属中有慢阻肺家族史。
5.居住在空气污染严重地区,尤其是二氧化硫等有害气体污染的地区。
6.长期从事接触粉尘、有毒有害化学气体、重金属颗粒等工作。
7.在婴幼儿时期反复患下呼吸道感染。
8.居住在气候寒冷、潮湿地区以及使用燃煤、木柴取暖。
9.维生素A缺乏或者胎儿时期肺发育不良。
10.营养状况较差,体重指数较低。
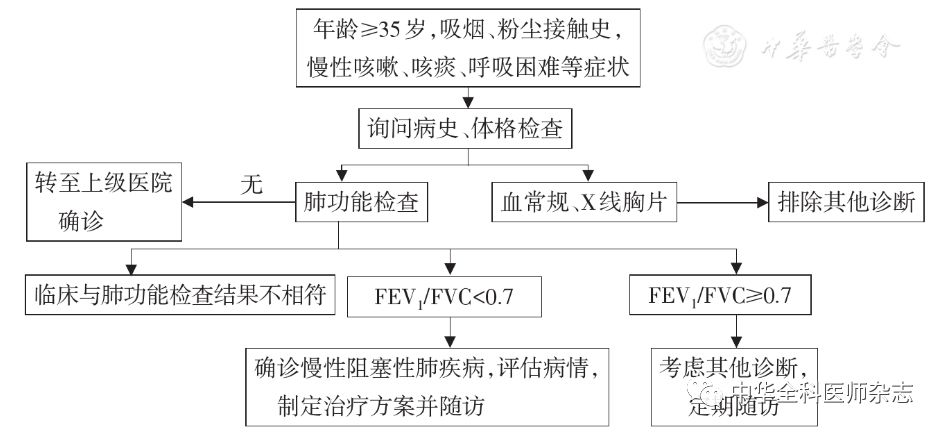
当基层医院不具备肺功能检查设备,临床医生可以通过问卷调查筛查慢阻肺高危人群(图1),对疑诊患者应该向上级医院转诊进一步确诊。
图1 慢性阻塞性肺疾病筛查问卷
(二)诊断
1.临床表现[31]:
(1)症状:多于中年发病,好发于秋冬寒冷季节。症状为慢性咳嗽、咳痰,少数可仅咳嗽不伴咳痰,甚至有明显气流受限但无咳嗽症状。痰为白色泡沫或黏液性,合并感染时痰量增多,转为脓痰。典型症状为气促或呼吸困难,早期仅于剧烈活动时出现,后逐渐加重,甚至发生于日常活动和休息时。晚期常有体重下降、食欲减退、精神抑郁和/或焦虑等,合并感染时可咳脓痰。后期出现低氧血症和/或高碳酸血症,可并发慢性肺源性心脏病和右心衰竭。
(2)体征:慢阻肺的早期体征可不明显,随着疾病进展,常出现以下体征:
①视诊:胸廓前后径增大,肋间隙增宽,剑突下胸骨下角增宽,称为桶状胸。部分患者呼吸变浅,频率增快,严重者可有缩唇呼吸等。
②触诊:双侧语颤减弱。
③叩诊:肺部过清音,心浊音界缩小,肺下界和肝浊音界下降。
④听诊:两肺呼吸音减弱,呼气期延长,部分患者可闻及湿性啰音和/或干性啰音,心音遥远,合并肺动脉高压时肺动脉瓣区第二心音(P2)较主动脉瓣区第二心音(A2)强(P2>A2)。
⑤肺外体征:低氧血症者可出现黏膜和皮肤发绀。伴二氧化碳潴留者可见球结膜水肿。伴有右心衰竭者可见下肢水肿和肝脏增大。
2.辅助检查[32]:
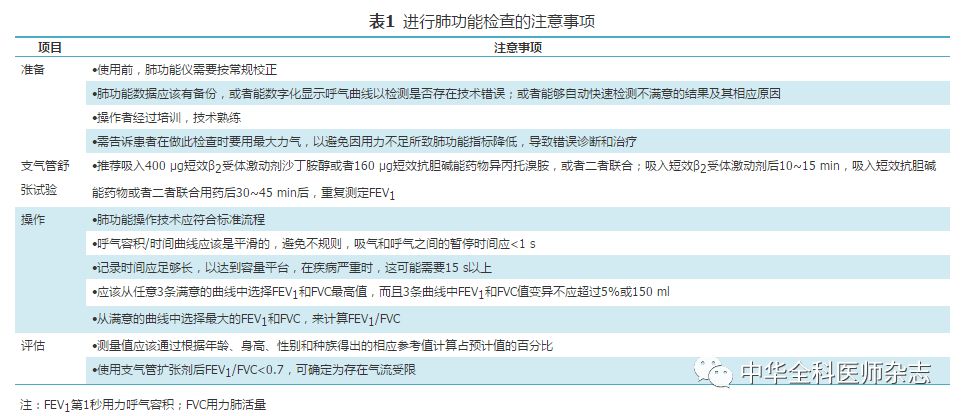
(1)肺功能检查:肺通气功能检查是判断气流受限的客观指标,重复性较好,对慢阻肺的诊断、严重程度评价、疾病进展、预后及治疗反应等均有重要意义。慢阻肺高危人群建议每年进行一次肺通气功能检测。气流受限是以FEV1占用力肺活量(forced vital capacity,FVC)百分比(FEV1/FVC)和FEV1占预计值%降低来确定的。FEV1/FVC是慢阻肺的一项敏感指标,可检出轻度气流受限。FEV1占预计值%是评价中、重度气流受限的良好指标,因其变异性小、易于操作,应作为慢阻肺的肺功能检查基本项目。患者吸入支气管扩张剂后的FEV1/FVC<0.7,可以确定为持续存在气流受限。单次支气管扩张剂后FEV1/FVC在0.6~0.8时,应重复肺功能检查以确诊[33]。因为在某些情况下,间隔一段时间后,由于个体差异,比值可能会发生改变。但对于支气管扩张剂后FEV1/FVC<0.6的慢阻肺患者,比值升至0.7以上的可能性不大。
气流受限可导致肺过度充气,使肺总量、功能残气量和残气容积增高,肺活量减低。肺总量增加不及残气容积增加的程度大,故残气容积与肺总量之比增高。肺泡隔破坏及肺毛细血管床丧失可使弥散功能受损,一氧化碳弥散量(DLCO)降低,DLCO与肺泡通气量之比较单纯DLCO更敏感。深吸气量是潮气量与补吸气量之和,深吸气量与肺总量之比是反映肺过度膨胀的指标,在反映慢阻肺呼吸困难程度甚至预测慢阻肺生存率方面具有意义。
支气管舒张试验作为辅助检查,与基础FEV1值及是否处于急性加重期和以往的治疗状态等有关,在不同时期检查结果可能不尽一致,因此要结合临床全面分析。需要指出的是,气流受限的可逆性程度不能作为区分慢阻肺与哮喘的唯一指标,也不能预测对支气管扩张剂或糖皮质激素长期治疗的反应性[15]。
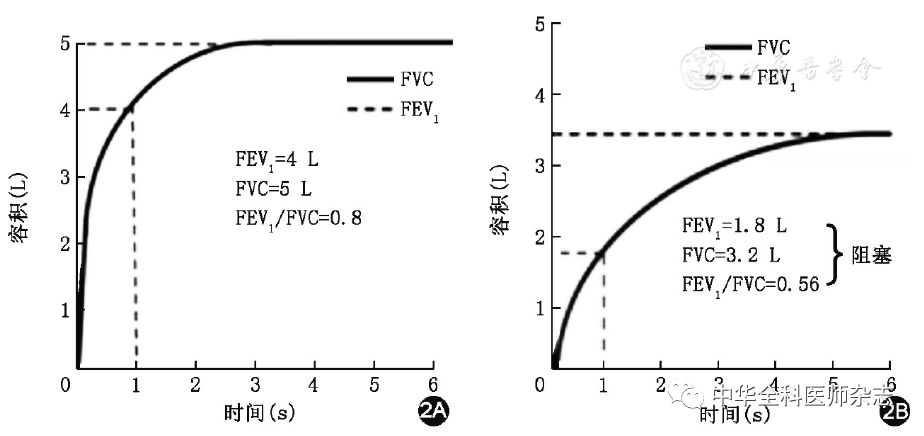
肺功能检查的注意事项[34,35]见表1,正常肺功能曲线和阻塞性肺疾病肺功能曲线[35]见图2。
注:FEV1第1秒用力呼气容积;FVC用力肺活量
图2肺功能曲线:正常肺功能曲线(2A);阻塞性肺疾病肺功能曲线(2B),慢性阻塞性肺疾病患者的典型表现为FEV1和FVC均出现下降
(2)胸部X线检查[15]:X线检查对确定肺部并发症及与其他疾病(如肺间质纤维化、肺结核等)鉴别具有重要意义。慢阻肺早期X线胸片可无明显变化,之后出现肺纹理增多和紊乱等非特征性改变;主要X线征象为肺过度充气,肺容积增大,胸腔前后径增长,肋骨走向变平,肺野透亮度增高,横膈位置低平,心脏悬垂狭长,肺门血管纹理呈残根状,肺野外周血管纹理纤细稀少等,有时可见肺大疱形成。并发肺动脉高压和肺源性心脏病时,除右心增大的X线特征外,还可有肺动脉圆锥膨隆,肺门血管影扩大及右下肺动脉增宽等。
(3)胸部CT检查[15]:CT检查一般不作为常规检查,但对于鉴别诊断具有重要价值。另外,高分辨率CT对辨别小叶中心型或全小叶型肺气肿及确定肺大疱的大小和数量,有很高的敏感性和特异性,对预计肺大疱切除或外科减容手术等的效果有一定价值。
(4)脉搏氧饱和度(SpO2)监测和血气分析[1]:慢阻肺稳定期患者如果FEV1占预计值%<40%,或临床症状提示有呼吸衰竭或右心衰竭时应监测SpO2。如果SpO2<92%,应进行血气分析检查。呼吸衰竭血气分析诊断标准为海平面呼吸空气时PaO2<60mmHg(1mmHg=0.133 kPa),伴或不伴有PaCO2>50mmHg。
(5)其他实验室检査[1]:低氧血症(PaO2<55mmhg)时血红蛋白和红细胞可以增高,血细胞比容>0.55可诊断为红细胞增多症。有的患者也可表现为贫血。合并感染时,痰涂片中可见大量中性粒细胞,痰培养可检出各种病原菌。
(三)诊断标准与诊断流程
慢阻肺的诊断应根据临床表现、危险因素接触史、体征及实验室检查等资料,综合分析确定。典型慢阻肺的诊断:呼吸困难、慢性咳嗽或咳痰;危险因素暴露史;肺功能检查吸入支气管扩张剂后FEV1/FVC<0.7提示气流受限,且除外其他疾病[1]。
基层医院医生应该仔细询问患者病史,遇到有慢性呼吸道症状,有危险因素暴露史,或有慢阻肺、哮喘等呼吸道疾病家族史者,应该进一步检查明确诊断。体格检查包括是否存在口唇、甲床发绀、颈静脉怒张、桶状胸、呼吸次数、呼吸音、啰音、心率、心律、双下肢浮肿、杵状指(趾)等[32]。
根据患者病情需要及医疗机构实际情况,恰当选择相应的检查项目,具体分为必做项目、推荐项目。必做项目包括血常规、肺通气功能检查(含支气管舒张试验)、X线胸片、心电图、经皮SpO2检测。根据当地医院条件和患者病情选择恰当检查或转诊到上级医院的推荐项目包括动脉血气分析、痰培养、胸部高分辨率CT检查、超声心动图、肺容量和弥散功能检查、6min步行距离(6MWD)、结核菌素纯蛋白衍生物(PPD)试验、D-二聚体、B型脑钠肽(BNP)或N-末端脑钠肽前体(NT-proBNP)、CRP、过敏原检测、总IgE、诱导痰细胞学分类、呼出气一氧化氮(FeNO)检测、双下肢静脉超声、肺通气灌注扫描或CT肺动脉造影(CTPA)、运动心肺功能等[32]。基层医院可使用图3的流程和标准进行慢阻肺诊断。
注:FEV1第1秒用力呼气容积;FV